Alibrandi S, Nicita F, Donato L, Scimone C, Rinaldi C, D'Angelo R, Sidoti A. Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes. Molecules. 2021 Nov 22;26(22):7045. doi: 10.3390/molecules26227045. PMID: 34834137; PMCID: PMC8618768.
For many years, trimethylaminuria (TMAU) has often been presented in fairly simple terms.
The traditional teaching is that someone either has "true genetic TMAU" or they do not. In this model, severe inherited TMAU is usually associated with two rare disease-causing mutations in the FMO3 gene (one inherited from each parent), resulting in a greatly reduced ability to convert trimethylamine (TMA) into the odourless trimethylamine N-oxide (TMAO).
This has influenced the typical clinical pathway.
If the urine test is positive, FMO3 genetic testing may be offered.
If the urine test is negative, investigation often stops, with no further genetic analysis.
While this approach identifies patients with classic severe TMAU, it may overlook a much larger group of people whose symptoms are intermittent, milder, or influenced by combinations of genetic and environmental factors.
The Missing Middle
In clinical practice, many patients who undergo FMO3 sequencing do not fit the textbook picture of carrying two rare pathogenic mutations.
Instead, they often carry combinations of common FMO3 variants that have traditionally been labelled as "benign" or "polymorphisms."
These combinations have generally attracted little attention because each individual variant appears to have only a small effect.
The important question is:
Can several mild variants together reduce FMO3 activity enough to contribute to TMAU symptoms?
This is exactly the question investigated by researchers at the University of Messina in Italy.
The Messina Study
Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes (2021)
The researchers analysed 26 patients with TMAU and found:
17 different FMO3 variants
26 different genetic haplotypes
many patients whose genetics did not fit the classic "two rare severe mutations" model
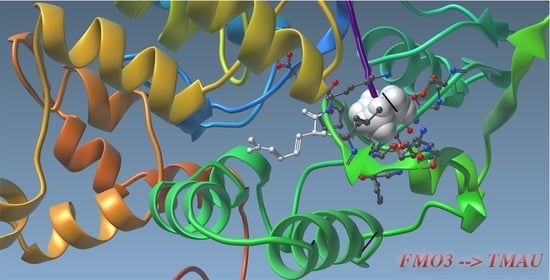
Using computational protein modelling, molecular docking and urine metabolite analysis, they proposed that combinations of variants (haplotypes) may alter how the FMO3 enzyme binds and processes trimethylamine.
Rather than viewing common variants individually, the paper suggests they should sometimes be considered together as functional genetic combinations.
The authors wrote:
"Variants classified as benign... have a high frequency in TMAU patients, frequently without the contemporary presence of causative mutations."
They therefore hypothesised that these variant combinations could reduce FMO3 activity sufficiently to contribute to disease.
Why Haplotypes Matter
A haplotype is simply a combination of genetic variants inherited together.
Each individual variant may have only a modest effect.
However, several small effects combined may reduce enzyme efficiency more than expected from considering each variant separately.
This idea is well recognised in many areas of genetics, where multiple low-impact variants together influence disease severity or susceptibility.
The Messina paper suggests that TMAU may follow a similar pattern in at least some patients.
Moving Beyond "Normal" or "Severe"
One way of thinking about this is to compare walking ability.
Traditional teaching effectively assumes there are only two categories:
walks normally
cannot walk
In reality, there is a broad spectrum.
Many people can walk but with limitations caused by arthritis, injuries, muscle weakness or neurological problems.
Likewise, FMO3 activity may exist on a spectrum rather than as a simple "working" versus "not working" enzyme.
Some people may have almost complete enzyme function.
Others may have very little.
Many may fall somewhere in between.
Those in this middle group might only develop symptoms during periods of illness, hormonal changes, dietary excess, stress, altered gut microbiota, or other metabolic challenges.
Why This Could Matter Clinically
The current diagnostic pathway often depends heavily on urine testing.
However, urinary TMA measurements can vary considerably depending on:
If a patient's urine test is normal on the day of testing, genetic analysis may never be performed.
If mild FMO3 haplotypes contribute to symptoms, this group could easily be under-recognised.
This remains a hypothesis rather than an established clinical fact, but it is one that deserves further investigation.
An Important Shift in Thinking
The significance of the Messina paper is not that it proves common variants cause TMAU.
Rather, it challenges the assumption that they are always clinically irrelevant.
Instead of asking:
"Does this patient have two rare pathogenic mutations?"
the question becomes:
"Could this particular combination of variants reduce FMO3 function enough to contribute to symptoms?"
That is a much more nuanced—and potentially more useful—way of approaching patients who do not fit the classic textbook description.
Since 2021
The Messina group's work has continued.
In 2024 they published a comprehensive review of TMAU covering genetics, molecular mechanisms, epidemiology and emerging treatments. The review again discusses the potential importance of variant combinations and the need to better understand how different FMO3 genotypes translate into clinical symptoms.
Research into treatments has also expanded.
A 2025 study investigated a mixture of postbiotics and tyndallized probiotics that reduced trimethylamine production in laboratory and animal models, highlighting growing interest in targeting the gut microbiome as part of TMAU management.
At the same time, other researchers continue to report newly identified pathogenic FMO3 mutations in different populations, demonstrating that classic severe genetic TMAU certainly exists while broadening our understanding of the condition worldwide.
If larger patient studies demonstrate that combinations of common FMO3 variants significantly reduce enzyme activity, it could eventually change how clinicians investigate patients whose symptoms are compatible with TMAU but who do not fit the traditional diagnostic model.
That would represent an important step toward recognising that TMAU may encompass a broader spectrum of FMO3 dysfunction than has historically been appreciated.
References
Alibrandi S, Nicita F, Donato L, Scimone C, Rinaldi C, D'Angelo R, Sidoti A. Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes. Molecules. 2021 Nov 22;26(22):7045. doi: 10.3390/molecules26227045. PMID: 34834137; PMCID: PMC8618768.
Alibrandi S, Nicita F, Donato L, et al. (2021). Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes.
Sidoti A, D'Angelo R, Castagnetti A, et al. (2024). Exploring Trimethylaminuria: Genetics and Molecular Mechanisms, Epidemiology, and Emerging Therapeutic Strategies.
Giannini G, Soldi S, Elli M, et al. (2025). A Mixture of Postbiotics/Tyndallized Probiotics Reduces Trimethylamine (TMA) in Trimethylaminuria Models.
Alghanem B, Alamri HS, Barhoumi T, et al. (2024). First Report from Saudi Arabia of Trimethylaminuria Caused by a Premature Stop Codon Mutation in the FMO3 Gene.